# 前言
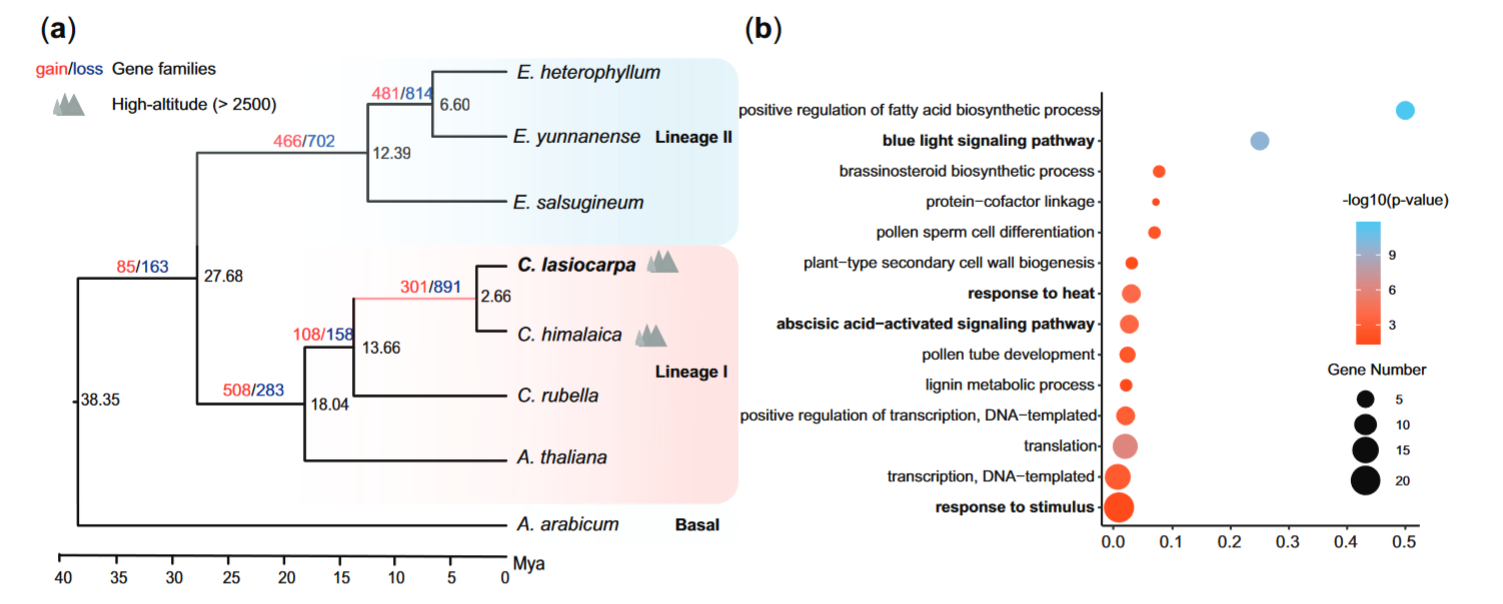
# 上篇推文中介绍到比较基因组学分析常用套路的第一步,利用单拷贝基因构建具有分化时间的物种树,补充一点,对于跨度较大的物种,可以选择单拷贝基因的方法,比如此次分析使用的物种。对于目级或者科级水平来讲,推荐使用共线性基因建树。以十字花科为例,如果用单拷贝基因,可能只有 1000 多组,但是使用共线性可能有接近 5000 组。共线性基因建树可以使用 WGDI 的方法,这部分内容我以后会探索一下。
![Sun et al 2022 WGDI]()
本篇推文主要讲基因家族的收缩与扩张分析,使用的软件是 cafe5,2020 年发表,相较于 cafe4 来讲操作更加方便并且新增了模型 (Gamma)
# 安装
git clone https://github.com/hahnlab/CAFE5.git | |
cd CAFE5 | |
./configure | |
make |
安装过程其实有点复杂,可能不同的服务器会出现不同的错误,这个请自行解决
# CAFE5 使用
输入文件至少要两个,一个是基因家族数目统计文件 Genefamilies_Count.tsv,一个是树文件 tree.txt(带有分化时间),还可以增加一个 lambda 文件
# 主要参数
--fixed_alpha, -a | |
Alpha value of the discrete gamma distribution to use in category calculations. If not specified, the alpha parameter will be estimated by maximum likelihood. | |
--lambda_per_family, -b | |
Estimate lambda by family (for testing purposes only). | |
--cores, -c | |
Number of processing cores to use, requires an integer argument. Default=All available cores. | |
--error_model, -e | |
Run with no file name to estimate the global error model file. This file can be provided in subsequent runs by providing the path to the Error model file with no spaces (e.g. -eBase_error_model.txt). | |
--Expansion, -E | |
Expansion parameter for Nelder-Mead optimizer, Default=2. | |
--rootdist, -f | |
Path to root distribution file for simulating datasets. | |
--help, -h | |
Help menu with a list of all commands. | |
--infile, -i | |
Path to tab delimited gene families file to be analyzed - Required for estimation. | |
--Iterations, -I | |
Maximum number of iterations that will be performed in lambda search. Default=300 (increase this number if likelihood is still improving when limit is hit). | |
--n_gamma_cats, -k | |
Number of gamma categories to use. If specified, the Gamma model will be used to run calculations; otherwise the Base model will be used. | |
--fixed_lambda, -l | |
Value (between 0 and 1) for a single user provided lambda value, otherwise lambda is estimated. | |
--log_config, -L | |
Turn on logging, provide name of the configuration file for logging (see example log.config file). | |
--fixed_multiple_lambdas, -m | |
Multiple lambda values, comma separated, must be used in conjunction with lambda tree (-y). | |
--output_prefix, -o | |
Output directory - Name of directory automatically created for output. Default=results. | |
--poisson, -p | |
Use a Poisson distribution for the root frequency distribution. If no -p flag is given, a uniform distribution will be used. A value can be specified (-p10, or --poisson=10); otherwise the distribution will be estimated from the gene families. | |
--pvalue, -P | |
P-value to use for determining significance of family size change, Default=0.05. | |
--chisquare_compare, -r | |
Chi square compare (not tested). | |
--Reflection, -R | |
Reflection parameter for Nelder-Mead optimizer, Default=1. | |
--simulate, -s | |
Simulate families. Either provide an argument of the number of families to simulate (-s100, or --simulate=100) or provide a rootdist file giving a set of root family sizes to match. Without such a file, the families will be generated with root sizes selected randomly between 0 and 100. | |
--tree, -t | |
Path to file containing newick formatted tree - Required for estimation. | |
--lambda_tree, -y | |
Path to lambda tree, for use with multiple lambdas. | |
--zero_root, -z | |
Include gene families that don't exist at the root, not recommended. |
其实主要用的就是 - i -p -k -y -t 这些参数
# 输入文件准备
# Genefamilies_Count.tsv
制表符分隔的基因家族计数文件,通常用 OrthoMCL,
OrthoFinder 等软件获取计数信息。
示例格式
Desc Family ID human chimp orang baboon gibbon macaque marmoset rat mouse cat horse cow | |
ATPase ORTHOMCL1 52 55 54 57 54 56 56 53 52 57 55 54 | |
(null) ORTHOMCL2 76 51 41 39 45 36 37 67 79 37 41 49 | |
HMG box ORTHOMCL3 50 49 48 48 46 49 48 55 52 51 47 55 | |
(null) ORTHOMCL4 43 43 47 53 44 47 46 59 58 51 50 55 | |
Dynamin ORTHOMCL5 43 40 43 44 31 46 33 79 70 43 49 50 | |
...... | |
.... | |
.. | |
DnaJ ORTHOMCL10016 45 46 50 46 46 47 46 48 49 45 44 48 |
我们首先利用 OrthoFinder 的 Orthogroups.GeneCount.tsv 文件生成符合要求的输入文件
cp Results_May02/Orthogroups/Orthogroups.GeneCount.tsv CAFE/ | |
awk 'OFS="\t" {$NF=""; print}' Orthogroups.GeneCount.tsv > tmp && awk '{print "(null)""\t"$0}' tmp > cafe.input.tsv && sed -i '1s/(null)/Desc/g' cafe.input.tsv && rm tmp |
查看文件格式
Desc Orthogroup Aof.pro Ath.pro Atr.pro Cba.pro Cri.pro Csa.pro Csu.pro Kle.pro Mpo.pro Nco.pro Osa.pro Ppa.pro Smo.pro Tpl.pro Vca.pro Vvi.pro Zma.pro | |
(null) OG0000000 145 112 95 5 372 129 3 1 2 217 126 16 206 419 4 177 117 | |
(null) OG0000001 9 4 3 1691 9 96 2 56 2 4 21 0 2 5 3 2 0 | |
(null) OG0000002 32 117 62 1 92 117 2 0 20 81 119 77 40 193 5 107 161 | |
(null) OG0000003 37 104 54 3 89 76 4 5 10 74 144 22 47 134 8 79 154 | |
(null) OG0000004 73 104 51 4 40 80 2 10 12 76 87 33 22 136 5 97 135 | |
(null) OG0000005 28 46 36 11 37 47 0 3 50 81 81 32 48 120 0 54 73 | |
(null) OG0000006 41 43 74 6 38 57 0 4 25 57 52 19 33 155 0 87 40 | |
(null) OG0000007 58 52 60 0 18 42 0 0 12 50 56 17 57 99 1 82 52 | |
(null) OG0000008 38 57 26 7 52 47 4 6 19 40 59 43 20 29 1 41 80 | |
(null) OG0000009 46 57 26 1 25 46 1 2 11 52 65 29 13 50 1 48 87 |
生成之后还需要剔除不同物种间拷贝数差异过大的基因家族,否则会报错,有内置脚本可以使用,我在运行的时候需要去掉第一行才能使用
python ~/soft/CAFE5/tutorial/clade_and_size_filter.py -i cafe.input.tsv -o gene_family_filter.txt -s |
笨方法
awk 'NR==1 || $3<100 && $4<100 && $5<100 && $6<100 && $7<100 && $8<100 && $9<100 && $10<100 && $11<100 && $12<100 && $13<100 && $14<100 && $15<100 && $16<100 && $17<100 && $18<100 && $19<100 {print $0}' cafe.input.tsv >gene_family_filter.txt |
最后的文件格式,保证第一行的物种名字与进化树的一致即可
Desc Orthogroup Aof Ath Atr Cba Cri Csa Csu Kle Mpo Nco Osa Ppa Smo Tpl Vca Vvi Zma | |
(null) OG0000020 26 37 23 4 35 28 0 1 24 28 43 24 27 47 0 35 42 | |
(null) OG0000021 49 41 31 7 30 31 8 2 7 26 49 15 11 31 0 36 45 | |
(null) OG0000022 27 25 31 0 27 34 2 1 23 25 46 18 27 44 1 39 45 | |
(null) OG0000024 37 40 27 0 22 30 1 11 9 33 38 18 25 43 0 37 39 | |
(null) OG0000029 28 26 23 2 24 25 1 2 5 32 34 31 17 35 1 30 40 | |
(null) OG0000030 23 30 16 1 23 27 1 1 27 26 26 16 15 49 1 28 35 | |
(null) OG0000031 28 36 26 3 27 23 8 1 3 17 37 10 18 38 3 34 28 | |
(null) OG0000032 18 16 25 0 24 19 0 5 4 25 36 6 38 49 1 38 35 | |
(null) OG0000033 19 17 20 0 12 16 4 6 18 35 42 4 23 39 3 45 28 | |
(null) OG0000035 17 37 17 8 28 24 2 2 5 30 41 8 6 26 2 32 37 | |
(null) OG0000036 22 15 17 3 22 19 7 12 13 20 24 20 30 38 3 35 22 | |
(null) OG0000039 14 27 36 0 34 24 0 2 2 12 41 4 47 13 0 37 26 | |
(null) OG0000040 15 30 9 1 19 35 0 2 11 25 26 19 12 48 0 39 27 |
# tree.txt
本步骤直接使用 mcmctree 生成的 FigTree.tre 文件修改一下即可使用
grep "UTREE 1 =" FigTree.tre | sed -E -e "s/\[[^]]*\]//g" -e "s/[ \t]//g" -e "/^$/d" -e "s/UTREE1=//" > tree.txt |
# 运行 CAFE5
cafe5 -i gene_family_filter.txt -t tree.txt -o out -c 1 | |
## 如果使用 Gamma 模型与泊松分布 | |
cafe5 -i gene_family_filter.txt -t tree.txt -o out -c 1 -k 2 -p ## 注意 - k 可以调,一般为 2-5 |
结果还是报错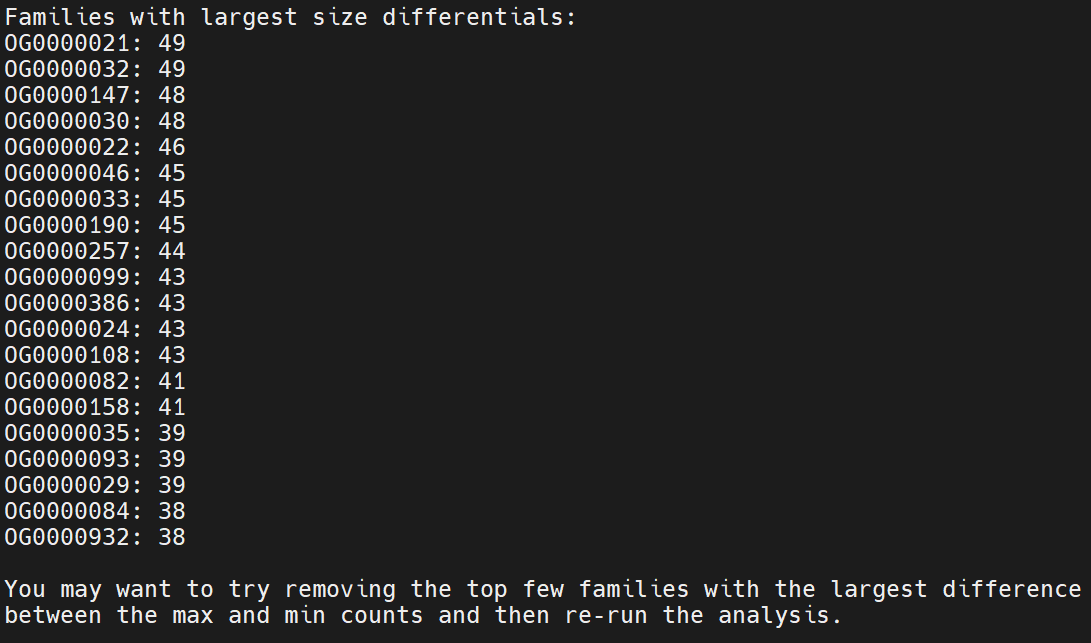
查了一下解决方法,可以将 lambda 值调低 (0.0001) 再进行计算
重新运行
cafe5 -i gene_family_filter.txt -t tree.txt -o out -c 16 -l 0.0001 -k 2 -p |
当大家出现这种错误时,可以尝试这种解决方案,单单删除这些家族是没用的。
# CAFE5 输出结果
# 结果文件
Gamma_asr.tre ## 每个基因家族的树文件 | |
Gamma_branch_probabilities.tab ## 每个分支计算的概率 | |
Gamma_category_likelihoods.txt | |
Gamma_change.tab ## 每一个基因家族在每个节点的收缩与扩增数目 | |
Gamma_clade_results.txt ## 每个节点基因家族的扩增 / 收缩数目 | |
Gamma_count.txt ## 每一个基因家族在每个节点的数目 | |
Gamma_family_likelihoods.txt | |
Gamma_family_results.txt ## 基因家族变化的 p 值和是否显著的结果 | |
Gamma_results.txt ## 模型,最终似然值,最终 Lambda 值等参数信息。 |
我们主要用的文件有 Gamma_asr.tre (主要对应后面表格中的节点)、Gamma_change.tab (看哪些基因家族在哪个节点发生变化)、Gamma_clade_results.txt (体现在树上,每个节点基因家族的收缩 / 扩增数目)、Gamma_family_results.txt (显著扩增 / 收缩的基因家族)
# 每个节点基因家族收缩 / 扩增数目的体现
其实有绘图脚本,但是很久没有更新,可能不适用于 CAFE5,我们可以自己画
将基因家族的扩增 / 收缩数目体现在树上,需要两个文件,Gamma_asr.tre,Gamma_clade_results.txt
cat Gamma_clade_results.txt | |
#Taxon_ID Increase Decrease | |
Mpo<21> 232 1298 | |
Ppa<20> 2231 371 | |
<31> 134 65 | |
<25> 949 220 | |
<23> 134 209 | |
Atr<13> 516 922 | |
Kle<29> 493 741 | |
<12> 245 56 | |
<28> 314 340 | |
<4> 118 176 | |
Cri<17> 1669 287 | |
<22> 214 184 | |
<19> 445 93 | |
<16> 291 352 | |
Osa<3> 579 572 | |
Aof<6> 935 840 | |
<8> 142 112 | |
Csa<1> 326 834 | |
<7> 640 138 | |
Tpl<15> 1147 395 | |
<14> 204 273 | |
Nco<11> 631 559 | |
Zma<2> 1776 232 | |
Vvi<5> 958 433 | |
<10> 413 112 | |
<9> 345 66 | |
Smo<18> 842 1315 | |
Cba<24> 744 1664 | |
<30> 23 17 | |
Ath<0> 1090 291 | |
Csu<27> 305 1560 | |
Vca<26> 438 1040 |
less Gamma_asr.tre | |
BEGIN TREES; | |
TREE OG0000021 = ((Kle<29>*_2:820.007,(((Mpo<21>*_7:428.285,Ppa<20>*_15:428.285)<23>_12:70.3982,((Cri<17>_30:404.796,(Tpl<15>_31:308.175,(Atr<13>_31:208.47,(Nco<11>*_26:176.909,(((Osa<3>*_49:45.2652,Zma<2>_45:45.2652)<7>_45:60.4652,Aof<6>*_49:105.73)<9>*_44:25.8965,(Vvi<5>_36:101.378,(Csa<1>*_31:83.0358,Ath<0>*_41:83.0358)<4>_36:18.3421)<8>_36:30.2489)<10>*_36:45.2824)<12>_31:31.5605)<14>_31:99.7053)<16>*_30:96.6205)<19>*_27:48.1939,Smo<18>_11:452.99)<22>*_13:45.694)<25>*_12:190.787,Cba<24>_7:689.47)<28>*_7:130.537)<31>_5:177.864,(Csu<27>*_8:874.03,Vca<26>*_0:874.03)<30>_5:123.841)<32>_5 |
可以看到少了个 32
我们可以利用 Gamma_change.tab 文件去找一下
经查看确实没有,忽略这一部分
将两个文件的 nodeid 对应即可绘图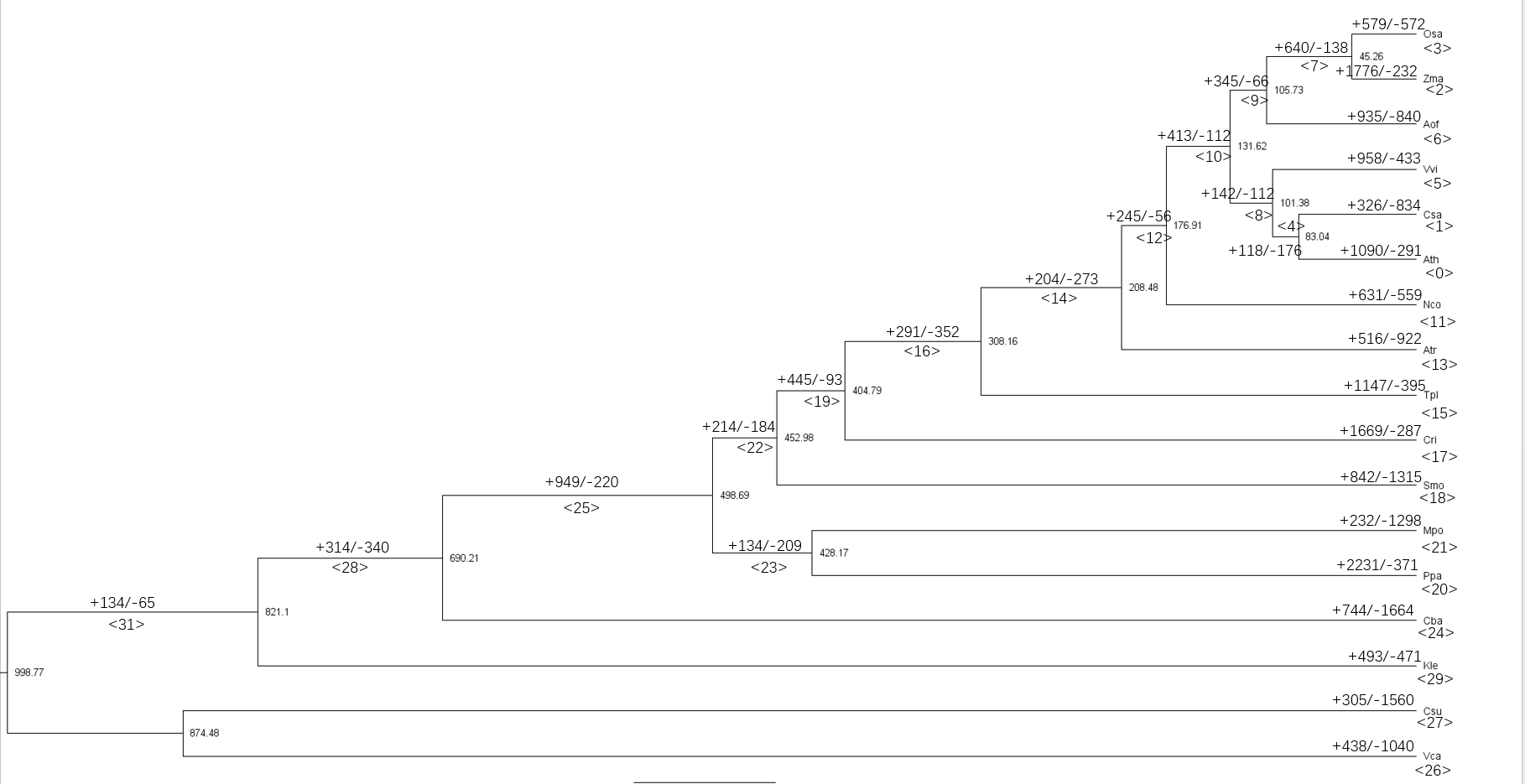
# 其他整理
相较于 CAFE4,这些结果并没有直接体现显著扩张 / 收缩的基因家族,或者我们想找一下某个节点具体扩张的基因,可以结合目前拿到的输出文件进行进一步整理
cat Gamma_family_results.txt |grep "y"|cut -f1 >p0.05.significant | |
# 提取显著扩张或收缩的 orthogroupsID | |
grep -f p0.05.significant Gamma_change.tab > Gamma_p0.05change.tab | |
# 显著扩张 / 收缩的基因家族在每个节点的收缩与扩增数目 | |
cat Gamma_p0.05change.tab | cut -f1,2 | grep "+[1-9]" | cut -f1 > node0significant.expand | |
#Ath 显著扩张的 orthogroupsID | |
wc -l node0significant.expand | |
#Ath 显著扩张的基因家族数目 | |
cp ../../Results_May02/Orthogroups/Orthogroups.tsv ./ | |
grep -f node0significant.expand Orthogroups.tsv | cut -f3 | sed "s//\n/g" | sed "s/\t/\n/g" | sed "s/,//g" | sort | uniq > node0significant.expand.genes | |
# 提取 Ath 显著扩张的基因,方法一 | |
cp ../../Results_May02/Orthogroups/Orthogroups.txt ./ | |
grep -f node0significant.expand Orthogroups.txt |sed "s//\n/g" | grep "Ath" | sort | uniq > node0significant.expand.genes | |
# 提取 Ath 显著扩张的基因,方法二 |
# 结语
总体来说,个人感觉 CAFE5 要比 CAFE4 方便很多,但是输出结果的可视化方面还需要加强,下一篇推文将简单实践某个节点的基因功能富集分析 (GO/KEGG), 类似于分析下图的红色节点。