# 前言
之前有写过单拷贝基因构建物种树的流程,现在我对流程进行优化,而且将会增加后续的基因家族收缩与扩张分析,希望对大家的分析有所帮助
# 物种选择
在此我不在介绍软件的原理和安装了,大家可以去看我之前的推文
物种选择此次包含了整个绿色植物,从绿藻门到被子植物
大部分基因组是在 jgi phytozome 上下载的
绿藻门 Chlorophyte:小球藻:Coccomyxa subellipsoidea (Csu), 团藻:Volvox carteri (Vca)
轮藻门 Charophyta:布氏轮藻:Chara braunii (Cba), Klebsormidium nitens (Kle)
苔藓植物门 Bryophyta:地钱:Marchantia polymorpha (Mpo), 小立碗藓:Physcomitrium patens (Ppa)
石松类 Lycopsida: 江南卷柏:Selaginella moellendorffii (Smo)
蕨类 Ferns: 水蕨:Ceratopteris richardii (Cri)
裸子植物门 Gymnospermae:北美乔柏 Thuja plicata (Tpl)
基部被子植物 Basial Angiosperm:无油樟:Amborella trichopoda (Atr), 睡莲:Nymphaea colorata (Nco)
双子叶植物 Eudicot:葡萄:Vitis vinifera (Vvi), 拟南芥:Arabidopsis thaliana (Ath), 黄瓜:Cucumis sativus (Csa)
单子叶植物 Monocot: 芦笋:Asparagus officinalis (Aof), 水稻:Oryza sativa (Osa), 玉米:Zea mays (Zma)
下载完成后我对每个蛋白序列的 ID 前面加上了物种简写,方便后续使用
sed -i "s/>/>Ath/g" Ath.pro.fasta
# Orthofinder 获得单拷贝基因
将所有物种的蛋白文件放到 CGA 文件夹下
orthofinder -t 16 -f CGA/ |
我们主要使用 Orthoogroups 查看正交群的基因和使用 Single_Copy_Orthologue_Sequences 里的单拷贝基因构建系统发育树
WorkingDirectory 其中包含运算过程的中间文件,例如 diamond 结果,如果我们想去掉某一物种,在 SpeciesIDs.txt 中将该物种注释掉
0: Aof.pro.fasta | |
1: Ath.pro.fasta | |
2: Atr.pro.fasta | |
3: Cba.pro.fasta | |
4: Cri.pro.fasta | |
5: Csa.pro.fasta | |
6: Csu.pro.fasta | |
7: Kle.pro.fasta | |
8: Mpo.pro.fasta | |
9: Nco.pro.fasta | |
10: Osa.pro.fasta | |
11: Ppa.pro.fasta | |
#12: Pum.pro.fasta | |
13: Smo.pro.fasta | |
14: Tpl.pro.fasta | |
15: Vca.pro.fasta | |
16: Vvi.pro.fasta | |
17: Zma.pro.fasta |
orthofinder -b WorkingDirectory |
如果想增加额外的物种进行分析,可以按照如下方式运行
orthofinder -b WorkingDirectory -f new_fasta_directory |
# 利用单拷贝基因构建系统发育树
Orthofinder 的 Single_Copy_Orthologue_Sequences 下存放着单拷贝同源基因的序列,我们可以利用这些序列构建系统发育树
建树方法可以分为串联法与并联法
不同的是,串联法是将得到的单拷贝同源基因比对后进行了串联,每个物种都得到一个很大的序列,然后进行建树;并联法是将每个单拷贝同源基因集比对后建树,然后再利用 Astral 构建了物种树,这里目前只讲串联法,并联法正在研究
# 串联法建树
vi test.sh | |
for i in *.fa | |
do | |
file=${i%.fa*} | |
mafft --maxiterate 1000 --localpair "${file}.fa" > "${file}.aln" ## 多序列比对 | |
(也可以添加一步,将蛋白文件转为 cds 文件再进行后续的分析,pal2nal.pl "${file}.aln" "${file}.cds" -output fasta > "${file}.cds.aln) | |
Gblocks "${file}.aln" -b4=5 -b5=h -t=p -e=.gb ## 提取保守序列 | |
seqkit seq "${file}.aln.gb" -w 0 > "${file}.new.aln" ## 多行序列并为一行 | |
awk '{if (NR%2==1) print substr ($1, 1, 4); else print $0}' "${file}.new.aln" > "${file}.final.aln" ## ID 修饰 | |
done |
这一步需要的软件 (mafft,Gblocks,seqkit) 请自行安装
接下来将所有的 aln 文件串联
seqkit concat *.final.aln > all.fa |
将所有单拷贝基因串联在一起后,进行模型预测,这次使用 modeltestng 进行预测
modeltest-ng -i all.fa -d aa -o modeltest -p 16 |
在 modeltest.out 文件中,我们可以看到我们需要的模型并且可以直接 copy 命令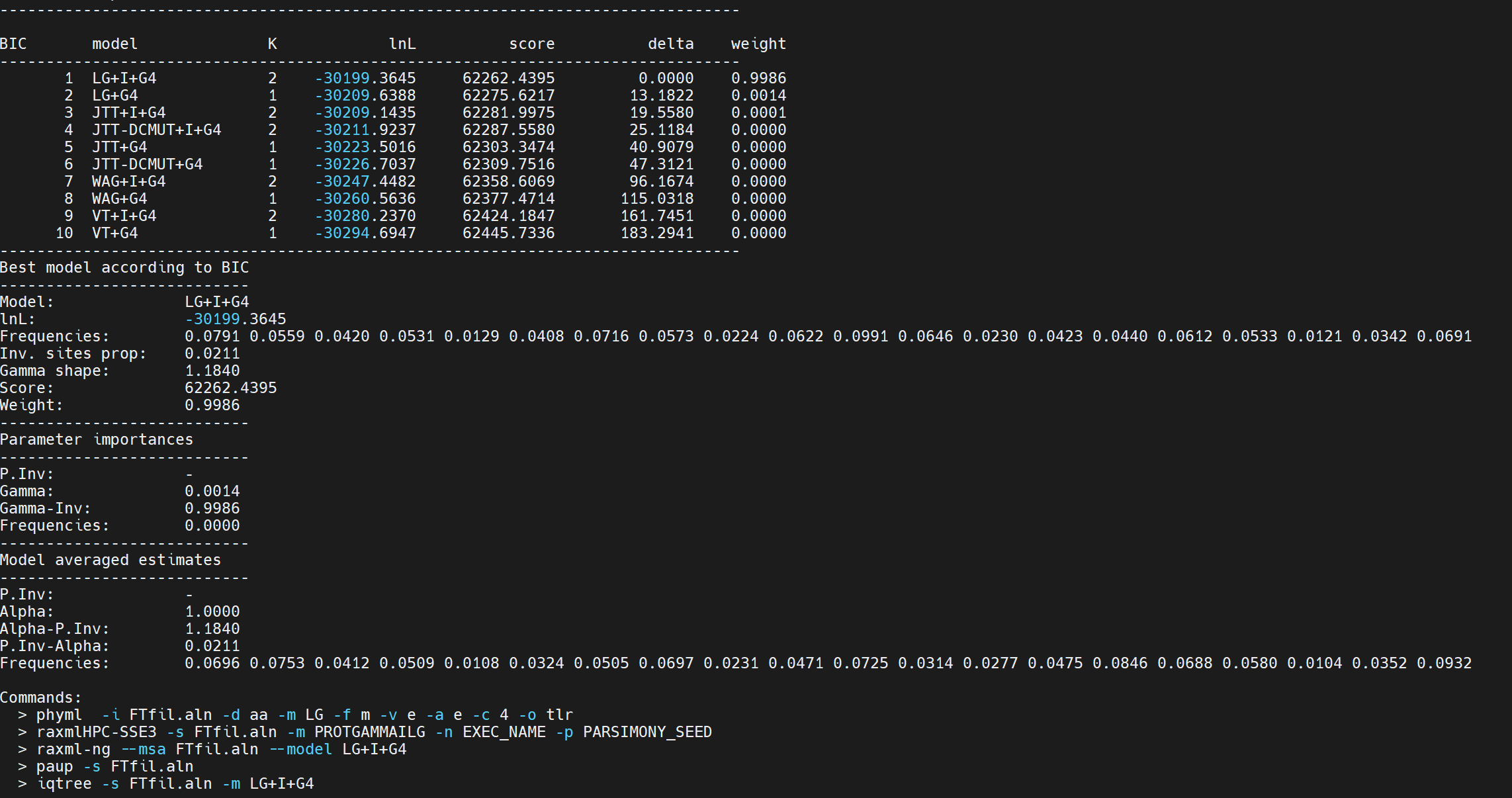
接下来进行建树
raxmlHPC-PTHREADS-SSE3 -T 32 -f a -x 123 -p 123 -N 10000 -m PROTGAMMAILGF -k -O -o Csu,Vca -n all.tre -s all.fa |
得到 RAxML_bipartitions.all.tre 文件查看树结构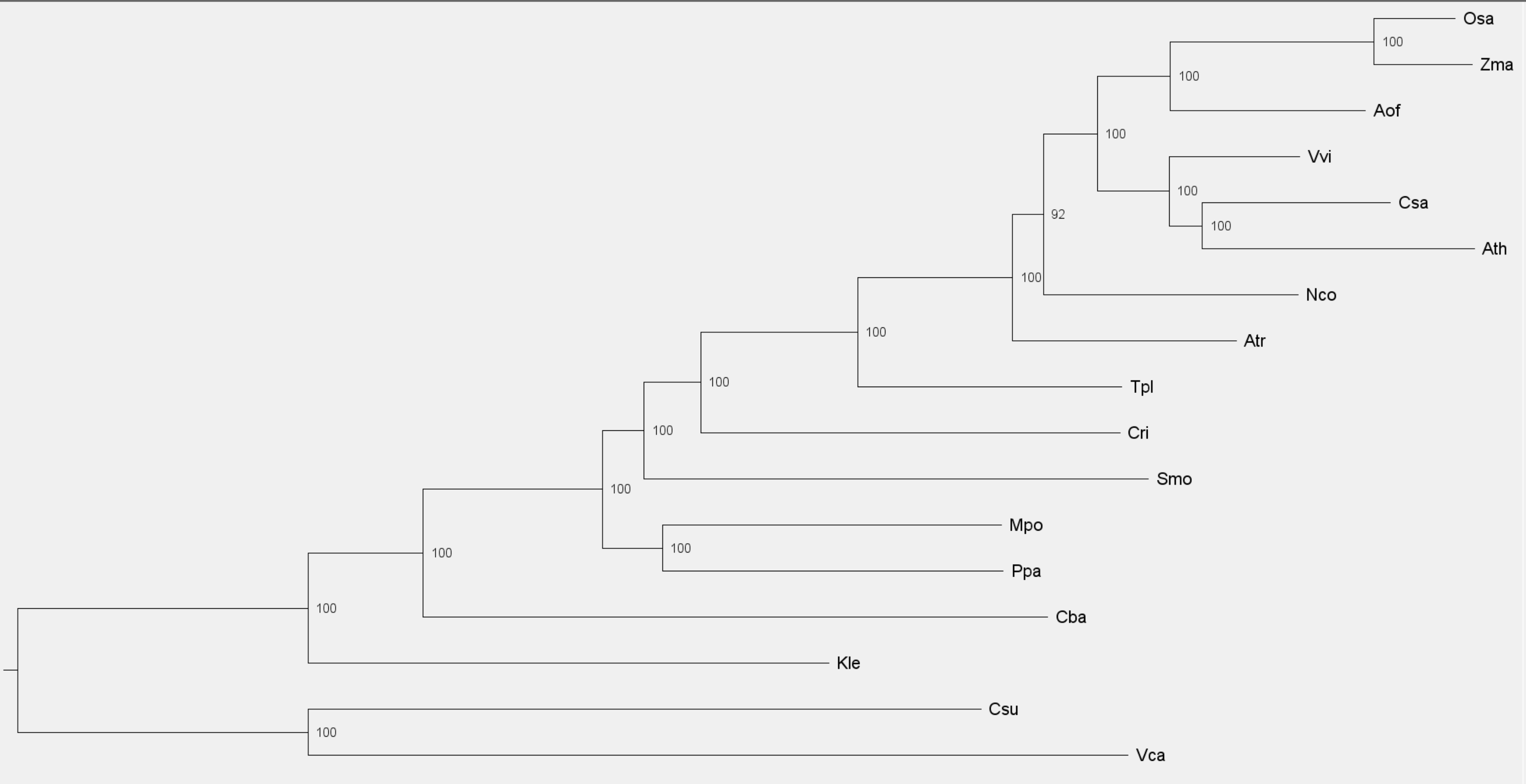
结果和进化关系是一致的
# 并联法建树
并联法建树使用 Astral,将所有的单拷贝基因树合并为一颗物种树
orthDir=~/CGApaper/protein/OrthoFinder/Results_May02 #基于 Orthofinder 结果 | |
cat $orthDir/Orthogroups/Orthogroups_SingleCopyOrthologues.txt |while read aa; do cat $orthDir/Gene_Trees/$aa\_tree.txt |awk '{print $0}' ;done > SingleCopy.trees | |
sed -r 's/([(,]{1}[A-Za-z]+)_[^:]+/\1/g' SingleCopy.trees > Astral_input.trees | |
java -jar ~/soft/ASTRAL-5.7.1/astral.5.7.1.jar -i Astral_input.trees -o Astral_output.tree -t 8 |
最后得到 Astral_output.tree,里面包含了不同拓扑结构的可能性,目前先到这一步
(Csu,(Vca,(Kle,(Cba,((Smo,(Cri,(Tpl,((Atr,Nco)'[q1=0.56;q2=0.24;q3=0.21]':0.3957605767189867,((Ath,(Vvi,Csa)'[q1=0.59;q2=0.24;q3=0.17]':0.47671301702525964)'[q1=0.9;q2=0.06;q3=0.04]':1.82319624265162,(Aof,(Zma,Osa)'[q1=0.98;q2=0.01;q3=0.01]':3.3756896221149395)'[q1=0.73;q2=0.11;q3=0.16]':0.8957898742947543)'[q1=0.59;q2=0.13;q3=0.28]':0.4793457920367108)'[q1=0.95;q2=0.01;q3=0.04]':2.471696418051194)'[q1=0.92;q2=0.02;q3=0.06]':2.040688555382602)'[q1=0.62;q2=0.18;q3=0.2]':0.5471348976356571)'[q1=0.37;q2=0.32;q3=0.32]':0.04487496622879318,(Ppa,Mpo)'[q1=0.74;q2=0.15;q3=0.1]':0.9286981407683164)'[q1=0.86;q2=0.07;q3=0.07]':1.5475625087160123)'[q1=0.56;q2=0.21;q3=0.23]':0.4056697116894409)'[q1=0.98;q2=0.02;q3=0.01]':3.121909101338843):0.0); |
# 物种分化时间计算
此次使用 mcmctree 计算物种的分化时间,首先在 timetree 上查询物种的分化时间进行标定,修改树文件,需要注意在标定时间是尽量较早的节点和较晚的节点都进行标定
17 1 | |
((Kle,(((Mpo,Ppa),((Cri,(Tpl,(Atr,(Nco,(((Osa,Zma)'>42<52',Aof)'>104<125',(Vvi,(Csa,Ath))'>98<117')'>85<128')'>173<199')'>136>247')'>289<330')'>392<422',Smo)'410<468')'>472<515',Cba)),(Csu,Vca)'>800<1000')'>725<1200'; |
配置文件书写
cp ~/soft/paml4.9i/mcmctree.ctl mcmctree1.ctl | |
vi mcmctree1.ctl | |
seed = -1 | |
seqfile = all.fa | |
treefile = all.tre | |
outfile = out | |
ndata = 1 ## 必须注意为 1 | |
seqtype = 2 * 0: nucleotides; 1:codons; 2:AAs ## 序列格式,蛋白就选择 AA | |
usedata = 3 * 0: no data; 1:seq like; 2:use in.BV; 3: out.BV ## 此步骤先设为 3 | |
clock = 3 * 1: global clock; 2: independent rates; 3: correlated rates | |
RootAge = <1.0 * safe constraint on root age, used if no fossil for root. | |
model = 0 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85 | |
alpha = 0 * alpha for gamma rates at sites | |
ncatG = 5 * No. categories in discrete gamma | |
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)? | |
BDparas = 1 1 0 * birth, death, sampling | |
kappa_gamma = 6 2 * gamma prior for kappa | |
alpha_gamma = 1 1 * gamma prior for alpha | |
rgene_gamma = 2 2 * gamma prior for overall rates for genes | |
sigma2_gamma = 1 10 * gamma prior for sigma^2 (for clock=2 or 3) | |
finetune = 1: 0.1 0.1 0.1 0.01 .5 * auto (0 or 1) : times, musigma2, rates, mixing, paras, FossilErr | |
print = 1 | |
burnin = 20000 | |
sampfreq = 2 | |
nsample = 100000 | |
*** Note: Make your window wider (100 columns) before running the program. |
之后运行
mcmctree mcmctree.ctl |
运行结果产生一个文件名为 out.BV,我们拷贝到两次分化时间估计的目录里,并用 in.BV 作为新的命名
cp out.BV ../02.Time/rep1/in.BV | |
cp out.BV ../02.Time/rep2/in.BV |
之后在运行两次重复
首先修改配置文件
seed = -1 | |
seqfile = all.fa | |
treefile = all.tre | |
outfile = out | |
ndata = 1 | |
seqtype = 2 * 0: nucleotides; 1:codons; 2:AAs | |
usedata = 2 * 0: no data; 1:seq like; 2:use in.BV; 3: out.BV | |
clock = 3 * 1: global clock; 2: independent rates; 3: correlated rates | |
RootAge = <1.0 * safe constraint on root age, used if no fossil for root. | |
model = 0 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85 | |
alpha = 0 * alpha for gamma rates at sites | |
ncatG = 5 * No. categories in discrete gamma | |
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)? | |
BDparas = 1 1 0 * birth, death, sampling | |
kappa_gamma = 6 2 * gamma prior for kappa | |
alpha_gamma = 1 1 * gamma prior for alpha | |
rgene_gamma = 2 2 * gamma prior for overall rates for genes | |
sigma2_gamma = 1 10 * gamma prior for sigma^2 (for clock=2 or 3) | |
finetune = 1: 0.1 0.1 0.1 0.01 .5 * auto (0 or 1) : times, musigma2, rates, mixing, paras, FossilErr | |
print = 1 | |
burnin = 800000 | |
sampfreq = 40 | |
nsample = 6500000 | |
*** Note: Make your window wider (100 columns) before running the program. |
最后我们会得到如下文件
all.fa all.phy all.tre FigTree.tre in.BV mcmctree.ctl mcmc.txt out SeedUsed |
检查两次结果的 FigTree.tre 文件,如果相差不大就可以使用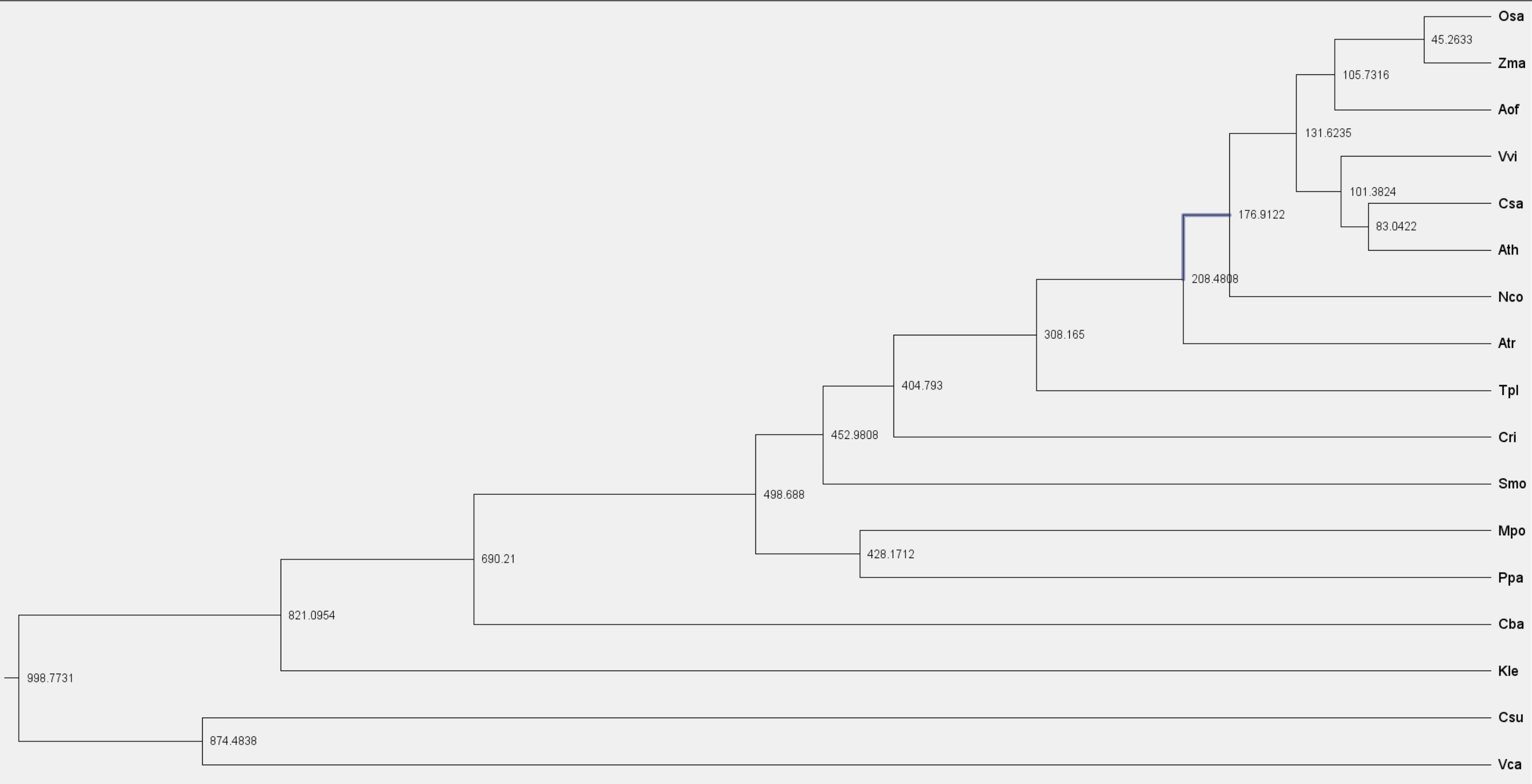
之后可以进行基因家族的收缩与扩张分析了,这部分之后在写