# 目录
1.Module 1 - Introduction to RNA sequencing
2.Module 2 - RNA-seq Alignment and Visualization
3.Module 3 - Expression and Differential Expression
4.Module 4 - Isoform Discovery and Alternative Expression
- Reference Guided Transcript Assembly
- de novo Transcript Assembly
- Transcript Assembly Merge
- Differential Splicing
- Splicing Visualization
5.Module 5 - De novo transcript reconstruction
6.Module 6 - Functional Annotation of Transcripts
# 2.4 Alignment Visualization
在我们可以在 IGV 浏览器中查看我们的结果之前,我们需要索引我们的 BAM 文件。为此,我们将使用 samtools 索引。为了方便以后,为所有 bam 文件建立索引。
cd align | |
find *.bam -exec echo samtools index {} \; | sh |
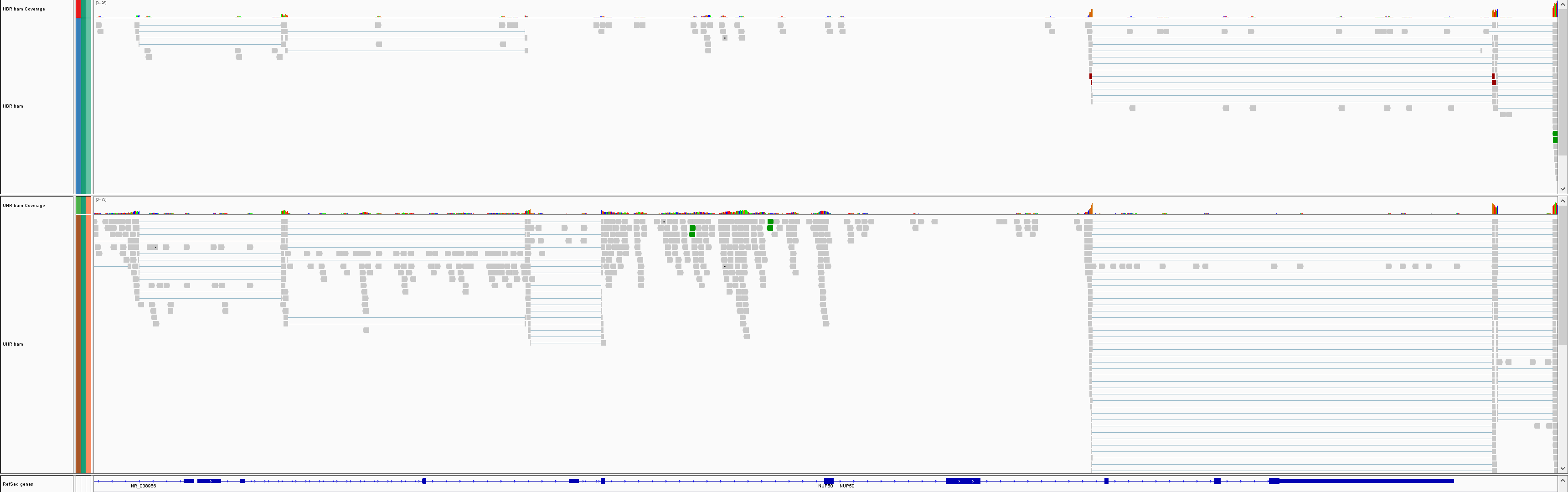
# 可视化比对结果
在 IGV 加载 UHR 与 HBR 的 bam 文件
# 练习:
尝试在 RNAseq 数据中找到一个变量位置:
- HINT: DDX17 is a highly expressed gene with several variants in its 3 prime UTR.
- Other highly expressed genes you might explore are: NUP50, CYB5R3, and EIF3L (all have at least one transcribed variant).
- Are these variants previously known (e.g., present in dbSNP)?
- How should we interpret the allele frequency of each variant? Remember that we have rather unusual samples here in that they are actually pooled RNAs corresponding to multiple individuals (genotypes).
- Take note of the genomic position of your variant. We will need this later.
# BAM read counting
首先,使用 samtools mpileup 来可视化相应的区域。
mkdir bam_readcount | |
cd bam_readcount | |
samtools mpileup -f ../chr22_only.fa -r 22:18918457-18918467 ../align/UHR.bam ../align/HBR.bam |
每一行由染色体、位点、碱基、覆盖位点的 reads 数、reads base 和 base 质量组成。在 read 碱基列,点表示与正链参考碱基匹配,逗号表示反链匹配,ACGTN 表示正链不匹配,ACGTN 表示反链不匹配。模式 +[0-9]+[ACGTNacgtn]+ 表示在这个参考位置和下一个参考位置之间有一个插入。插入的长度由模式中的整数给出,然后是插入的序列。有关输出的更多解释,请参阅 samtools pileup/mpileup 文档
- http://samtools.sourceforge.net/pileup.shtml
- http://samtools.sourceforge.net/mpileup.shtml
现在,使用 bam-readcount 来计数。首先,创建一个 bed 文件,其中包含一些感兴趣的位置 (我们将创建一个名为 snvs 的文件。使用 echo 命令 bed)。
它将包含一个单行,指定 chr22 上的变量位置,例如。
echo "22 38483683 38483683" | |
echo "22 38483683 38483683" > snvs.bed |
在这个列表上运行 bam-readcount 查看肿瘤和正常合并的 bam 文件
bam-readcount -l snvs.bed -f ../chr22_only.fa ../align/HBR.bam 2>/dev/null 1>HBR_bam-readcounts.txt | |
bam-readcount -l snvs.bed -f ../chr22_only.fa ../align/UHR.bam 2>/dev/null 1>UHR_bam-readcounts.txt |
从这个输出中,可以解析每个碱基的 reads 计数
cat UHR_bam-readcounts.txt | perl -ne '@data=split("\t", $_); @Adata=split(":", $data[5]); @Cdata=split(":", $data[6]); @Gdata=split(":", $data[7]); @Tdata=split(":", $data[8]); print "UHR Counts\t$data[0]\t$data[1]\tA: $Adata[1]\tC: $Cdata[1]\tT: $Tdata[1]\tG: $Gdata[1]\n";' | |
UHR Counts 22 38483683 A: 163 C: 0 T: 0 G: 163 | |
cat HBR_bam-readcounts.txt | perl -ne '@data=split("\t", $_); @Adata=split(":", $data[5]); @Cdata=split(":", $data[6]); @Gdata=split(":", $data[7]); @Tdata=split(":", $data[8]); print "HBR Counts\t$data[0]\t$data[1]\tA: $Adata[1]\tC: $Cdata[1]\tT: $Tdata[1]\tG: $Gdata[1]\n";' | |
HBR Counts 22 38483683 A: 75 C: 0 T: 0 G: 131 |