对于 RNA-seq 数据,有两种使用策略,一种是使用 HISAT2 + StringTie 先比对再组装,一种是从头组装,然后使用 PASA 将转录本比对到基因组上。在本篇教程中,只使用有参组装
参考链接:如何对基因组注释
首先,使用 HISAT2 将 RNA-seq 数据比对到参考基因组,这一步和之前相似,但是要增加一个参数 --dta ,使得 StingTie 能更好的利用双端信息
hisat2-build LH.mask.fa index/chi_masked ## 这个是屏蔽重复序列后的一个待发表基因组,与上两篇推文的基因组不一样 | |
hisat2 --dta -p 20 -x index/chi_masked -1 1-1_R1.fastq -2 1-1_R2.fastq | samtools sort -@ 10 > results/1-1.bam & | |
hisat2 --dta -p 20 -x index/chi_masked -1 1-2_R1.fastq -2 1-2_R2.fastq | samtools sort -@ 10 > results/1-2.bam & | |
hisat2 --dta -p 20 -x index/chi_masked -1 1-3_R1.fastq -2 1-3_R2.fastq | samtools sort -@ 10 > results/1-3.bam & | |
samtools merge -@ 10 results/merged.bam results/1-1.bam results/1-2.bam results/1-3.bam |
然后用 StringTie 进行转录本预测
stringtie -p 10 -o results/merged.gtf results/merged.bam |
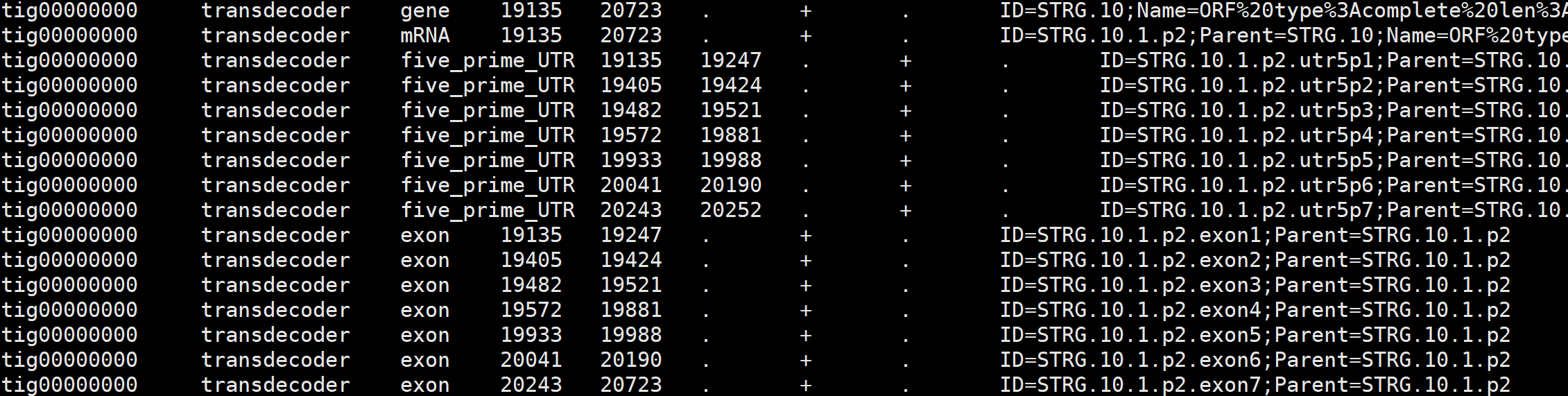
对于后续的 EvidenceModeler 而言,它不需要 UTR 信息,只需要编码区 CDS,需要用 TransDecoder 进行编码区预测
TransDecoder-TransDecoder-v5.5.0/util/gtf_genome_to_cdna_fasta.pl merged.gtf input/chi_masked.fa > transcripts.fasta | |
TransDecoder-TransDecoder-v5.5.0/util/gtf_to_alignment_gff3.pl merged.gtf > transcripts.gff3 | |
TransDecoder.LongOrfs -t transcripts.fasta | |
TransDecoder.Predict -t transcripts.fasta | |
TransDecoder-TransDecoder-v5.5.0/util/cdna_alignment_orf_to_genome_orf.pl \ | |
transcripts.fasta.transdecoder.gff3 \ | |
transcripts.gff3 \ | |
transcripts.fasta > transcripts.fasta.transdecoder.genome.gff3 |
最后结果 transcripts.fasta.transdecoder.genome.gff3 用于提供给 EvidenceModeler