参考链接:
https://github.com/velocyto-team/velocyto.R
http://velocyto.org/velocyto.py/index.html
http://pklab.med.harvard.edu/velocyto/notebooks/R/chromaffin2.nb.html
https://htmlpreview.github.io/?https://github.com/satijalab/seurat-wrappers/blob/master/docs/velocity.html
https://github.com/velocyto-team/velocyto.R/issues/16
https://www.jianshu.com/p/bce19672879e
# 目录
RNA 速率:使用 Seurat 的结果做 RNA velocity
# velocyto 下载
首先是下载 velocyto 生成 loom 文件
## 1. 创建 python>3.6 的环境 | |
conda create -n velocyto python=3.6 | |
## 2. 安装前置软件 | |
conda install numpy scipy cython numba matplotlib scikit-learn h5py click | |
pip install pysam | |
## 3. 安装 velocyto | |
pip install velocyto | |
## 4. 测试 | |
velocyto --help | |
Usage: velocyto [OPTIONS] COMMAND [ARGS]... | |
Options: | |
--version Show the version and exit. | |
--help Show this message and exit. | |
Commands: | |
run Runs the velocity analysis outputting a loom file | |
run10x Runs the velocity analysis for a Chromium Sample | |
run-dropest Runs the velocity analysis on DropEst preprocessed data | |
run-smartseq2 Runs the velocity analysis on SmartSeq2 data (independent bam file per cell) | |
tools helper tools for velocyto |
# repeat_masker.gtf 生成
运行 velocyto 需要准备三个文件,单细胞数据分析的结果文件,基因组注释文件,重复序列注释文件,其中前两个在单细胞分析时就会得到,关键是这个 repeat_masker.gtf
本人是做植物的,所以本次教程主要关注植物类 repeat_masker.gtf 的获得,人和小鼠的重复序列文件比较好得到,植物类首先可以看一下 Phtozome 数据库上想要研究的物种的注释文件夹下有没有 reapeat.gtf,没有就要我们自己生成了
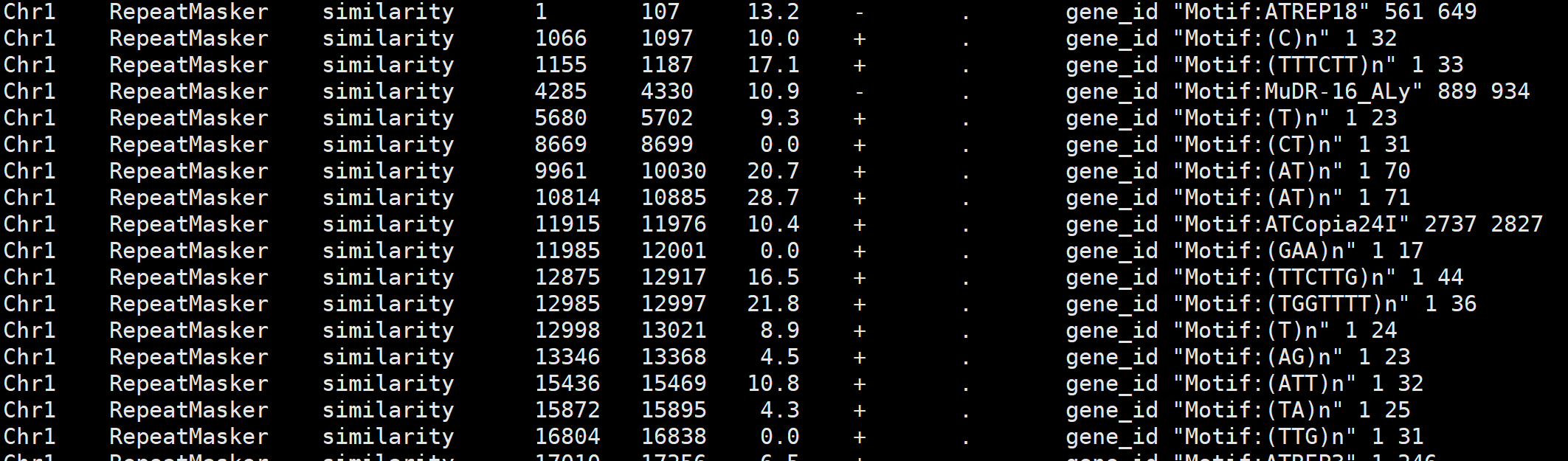
重复序列的注释文件我们一直没怎么关注过,对于做过基因组注释的童鞋来说,大家都忽略了一步,其实 repeatmasker 就可以生成重复序列的注释文件。
RepeatMasker -e ncbi -species arabidopsis -pa 40 -gff TAIR10.fa |
生成 gff 文件后,可以看到重复序列的点位以及属性,velocyto 主要使用的就是位点
# loom 文件生成
接下来是生成 loom 文件,运行 velocyto 需要准备三个文件,基因组注释文件 (gtf),repeat_masker.gtf (重复序列注释文件),cellranger 的结果文件夹 (以样本名 WT_1 为例,里面包含 cell matrix 和 bam 文件)
velocyto -m TAIR10_masked.gtf WANG/ TAIR10.gtf |
运行结束后会在 WANG 文件夹下生成 velocyto 文件夹,里面有 velocyto.loom 的文件,可以用于下一步的分析