生信技能树单细胞数据挖掘笔记 (2):创建 Seurat 对象并进行质控、筛选高变基因并可视化
生信技能树单细胞数据挖掘笔记 (4):其他分析(周期判断、double 诊断、细胞类型注释)
# 轨迹分析
通过构建细胞间的变化轨迹来重塑细胞随时间的变化过程。
##4、拟时轨迹分析 ---- | |
library (monocle) | |
data <- as (as.matrix (scRNA@assays$RNA@counts), 'sparseMatrix') | |
# count 矩阵 | |
pd <- new ('AnnotatedDataFrame', data = scRNA@meta.data) | |
# meta 表转成特定格式 | |
fData <- data.frame (gene_short_name = row.names (data), row.names = row.names (data)) | |
fd <- new ('AnnotatedDataFrame', data = fData) | |
# 基因名表转成特定格式 | |
mycds <- newCellDataSet (data, | |
phenoData = pd, | |
featureData = fd, | |
expressionFamily = negbinomial.size ()) | |
#expressionFamily 参数用于指定表达矩阵的数据类型,有几个选项可以选择: | |
# 稀疏矩阵用 negbinomial.size (),FPKM 值用 tobit (),logFPKM 值用 gaussianff () | |
##save (mycds, file = "../../tmp/mycds_raw.Rdata") | |
##rm (list = ls ()) | |
##load ("../../tmp/mycds_raw.Rdata") | |
#library ("monocle") | |
mycds <- estimateSizeFactors (mycds) | |
mycds <- estimateDispersions (mycds, cores=4, relative_expr = TRUE) #! | |
# 完成数据导入和预处理后,就可以考虑选择特定基因代表细胞的发育特征 |
# 读入 maker 基因
mycds <- estimateSizeFactors (mycds) | |
mycds <- estimateDispersions (mycds, cores=4, relative_expr = TRUE) #! | |
# 完成数据导入和预处理后,就可以考虑选择特定基因代表细胞的发育特征 | |
# 这里可以选取我们之前挑选的 marker gene | |
load ("../markergene.Rdata") | |
markers.gene <- all.markers$gene | |
mycds <- setOrderingFilter (mycds, markers.gene) | |
# 降维 | |
mycds <- reduceDimension (mycds, max_components = 2, method = 'DDRTree') #! | |
# 耗时,耗内存 |
# 细胞排序
#降维 | |
mycds <- reduceDimension (mycds, max_components = 2, method = 'DDRTree') #! | |
# 耗时,耗内存 | |
# 排序 | |
mycds <- orderCells (mycds) | |
#save (mycds,file = "../../tmp/mycds_reduced.Rdata") | |
load ("../../tmp/mycds_reduced.Rdata") | |
p1 <- plot_cell_trajectory (mycds, color_by = "seurat_clusters") | |
ggsave ("../../out/3.4trajectory_1.pdf", plot = p1) | |
p2 <- plot_cell_trajectory (mycds, color_by = "State") | |
ggsave ("../../out/3.4trajectory_2.pdf", plot = p2) |
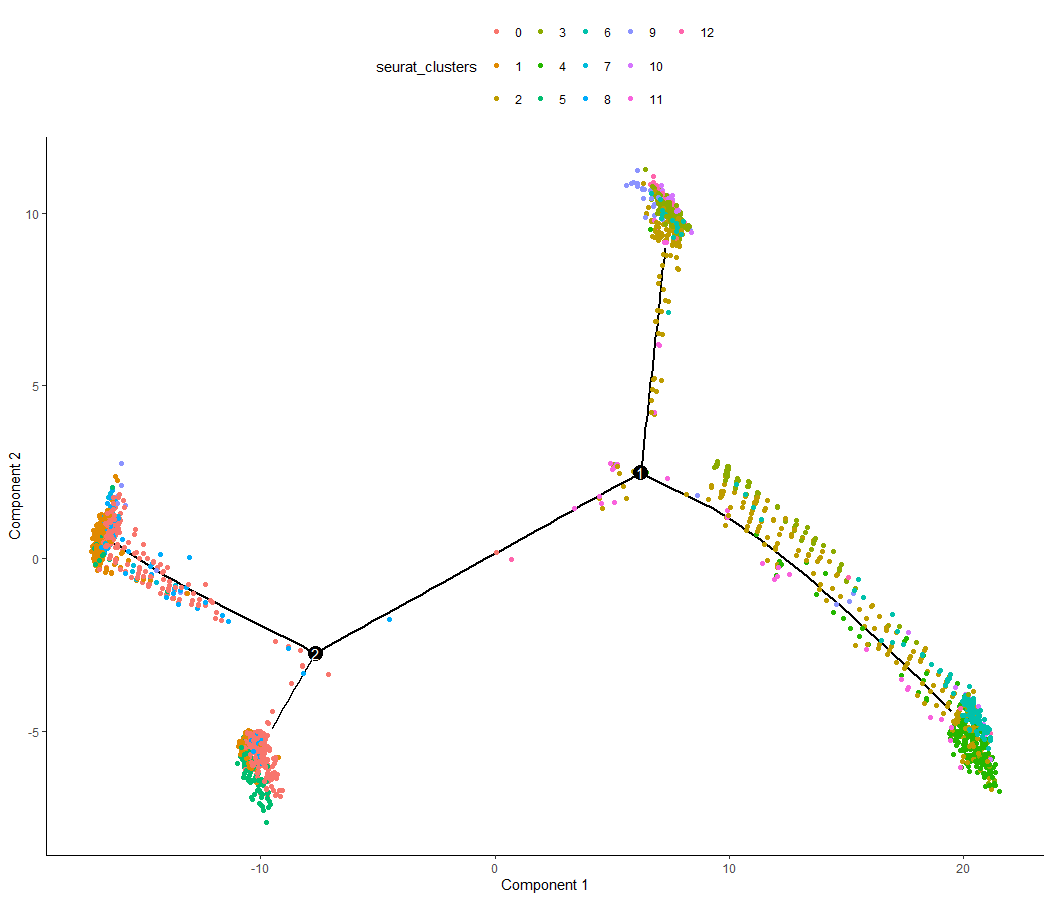
<center>
by_cluster
</center>
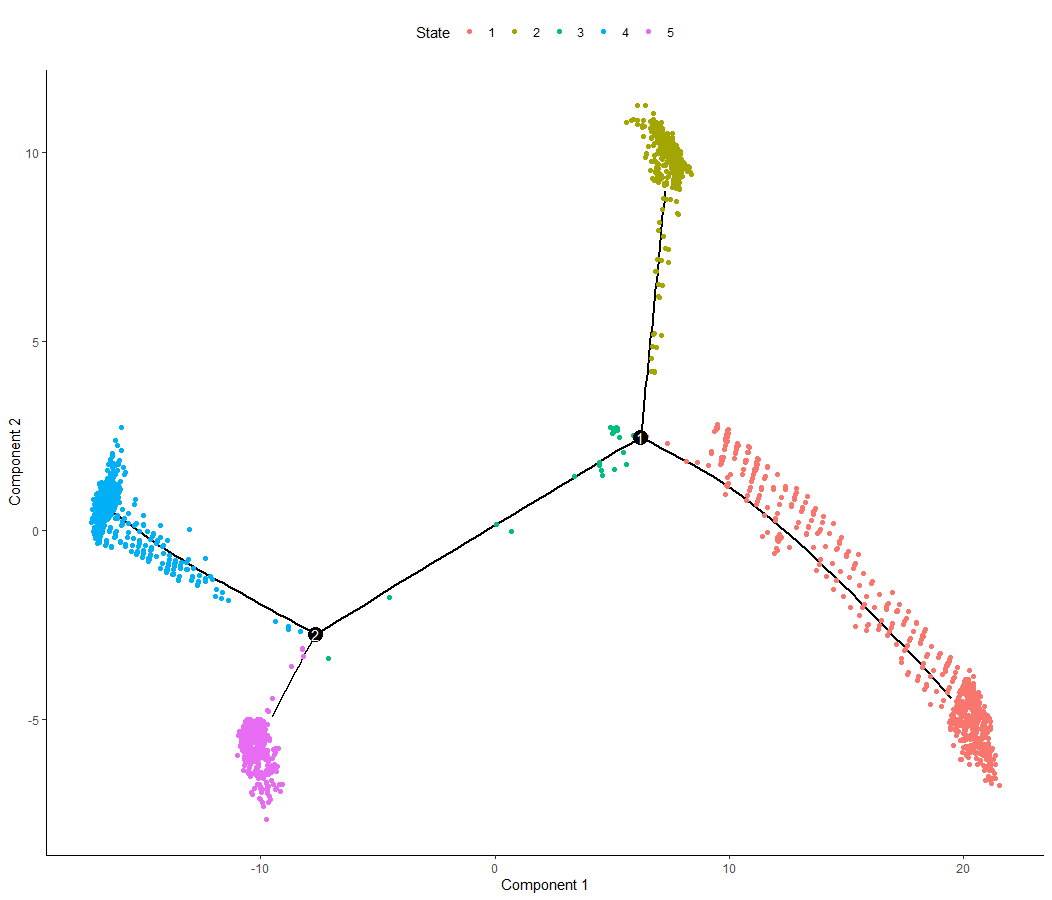
<center>
by_state
</center>