生信技能树单细胞数据挖掘笔记 (2):创建 Seurat 对象并进行质控、筛选高变基因并可视化
生信技能树单细胞数据挖掘笔记 (4):其他分析(周期判断、double 诊断、细胞类型注释)
# QC 之 double droplet 和周期判断
load ("../scRNA.Rdata") | |
library (Seurat) | |
##1、detect special cells---- | |
#empty droplet | |
##BiocManager::install ("DropletUtils") | |
library (DropletUtils) | |
e.out <- emptyDrops (GetAssayData (scRNA,slot="counts",assay="RNA")) | |
#Error in testEmptyDrops (m, lower = lower, ...) : | |
#no counts available to estimate the ambient profile | |
##https://support.bioconductor.org/p/123554/#123562 | |
# 如上回答所说,empty droplet 往往在第一步就已经过滤掉了,而一般上传到 GEO 的也都是过滤掉空液滴的。 | |
#double droplet | |
#https://osca.bioconductor.org/doublet-detection.html | |
#BiocManager::install ("scran") | |
head (scRNA@meta.data) | |
library (scran) | |
#GetAssayData (scRNA,slot="counts",assay="RNA")[1:8,1:4] | |
?doubletCluster #检查有无 double droplet 聚在一起的类 | |
db.test <- doubletCluster (GetAssayData (scRNA,slot="counts",assay="RNA"), | |
clusters=scRNA@meta.data$seurat_clusters) | |
head (db.test) | |
table (scRNA@meta.data$seurat_clusters) | |
library (scater) | |
chosen.doublet <- rownames (db.test)[isOutlier (db.test$N, | |
type="lower", log=TRUE)] | |
chosen.doublet #结果显示没有 | |
# 还有其它多种方法 |
# 周期诊断
##2、周期判断 ---- | |
# 在挑选 hvg gene 那一步因,可能会找到一些细胞周期相关基因; | |
# 它们会导致细胞聚类发生一定的偏移,即相同类型的细胞在聚类时会因为细胞周期的不同而分开。 | |
# 因此有必要查看是否有细胞周期相关基因的存在;若有,则剔除 | |
# 细胞周期有关基因 | |
?cc.genes | |
head (c (cc.genes$s.genes,cc.genes$g2m.genes)) | |
length (c (cc.genes$s.genes,cc.genes$g2m.genes)) | |
# [1] "MCM5" "PCNA" "TYMS" "FEN1" "MCM2" "MCM4" | |
# 查看我们选择的高变基因中有哪些细胞周期相关基因,及打分 | |
CaseMatch (c (cc.genes$s.genes,cc.genes$g2m.genes),VariableFeatures (scRNA)) | |
# 在 scRNA@meta.data 中添加 S.Score、G2M.Score 和 Phase 三列有关细胞周期的信息。 | |
g2m_genes = cc.genes$g2m.genes | |
g2m_genes = CaseMatch (search = g2m_genes, match = rownames (scRNA)) | |
s_genes = cc.genes$s.genes | |
s_genes = CaseMatch (search = s_genes, match = rownames (scRNA)) | |
scRNA <- CellCycleScoring (object=scRNA, g2m.features=g2m_genes, s.features=s_genes) | |
head (scRNA@meta.data) | |
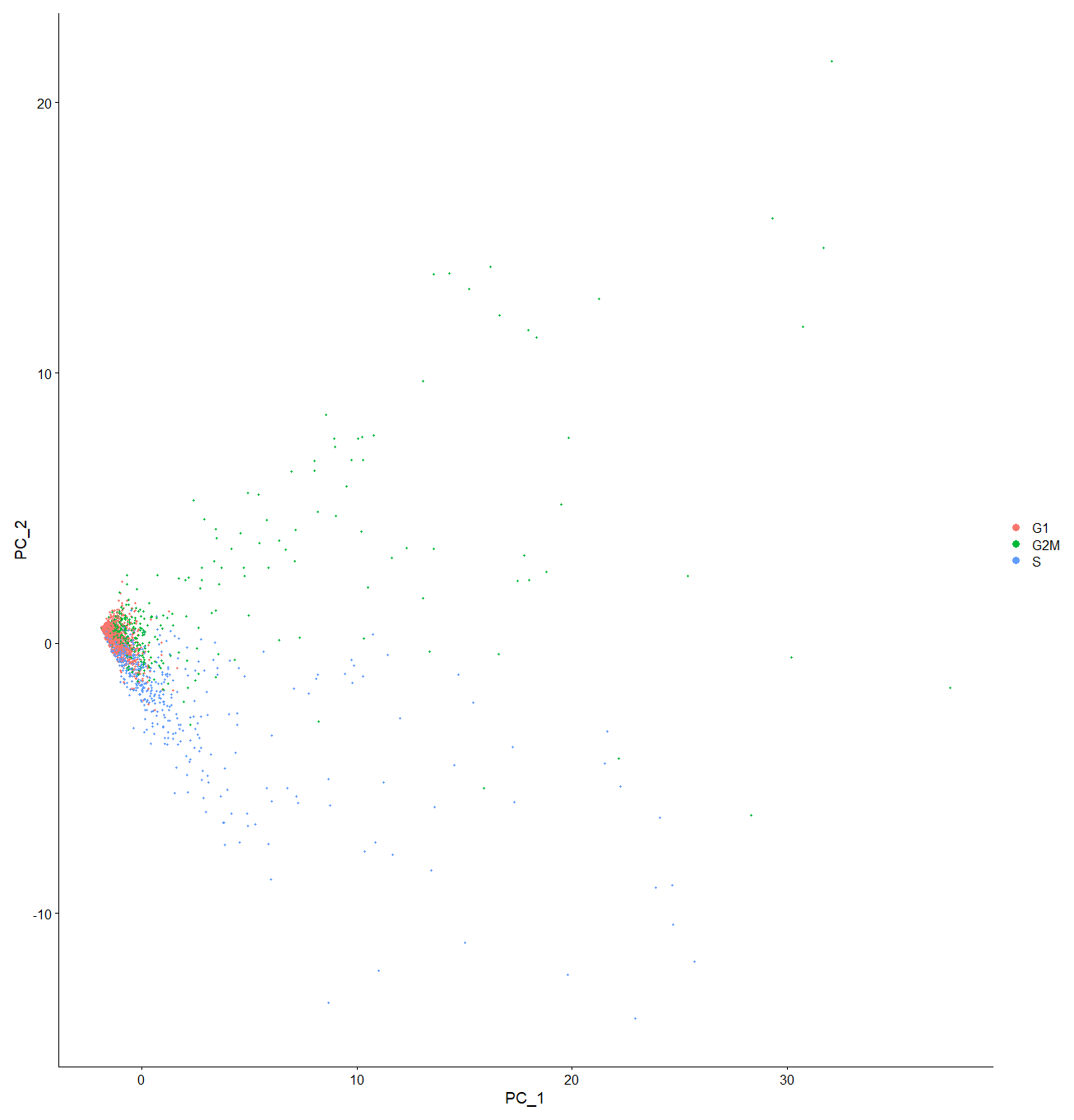
# 观察细胞周期相关基因是否影响聚类 | |
scRNA <- RunPCA (scRNA, features = c (s_genes, g2m_genes)) | |
p1 <- DimPlot (scRNA, reduction = "pca", group.by = "Phase") | |
ggsave ("../../out/3.2cell-cycle.pdf", plot = p1) | |
# 影响不大,基本重合在一起了 |
<center>
周期的影响并不大
</center>
# 单细胞类型注释
##3、cell annotation----- | |
# 对肿瘤细胞来说,分群后的细胞亚群注释是不可行的 | |
# 这里仅仅是演示 SingleR 做 cell annotation 的流程 | |
library (SingleR) | |
refdata <- get (load ("../../rawdata/HumanPrimaryCellAtlasData.Rdata")) | |
assay (refdata)[1:4,1:4] | |
head (refdata@colData) | |
head (refdata) | |
ref <- HumanPrimaryCellAtlasData () | |
# 参考数据库,等待时间较长。建议下载成功后,储存为 Rdata,以后方便使用。 | |
testdata <- GetAssayData (scRNA, slot="data") | |
# 参考数据库,等待时间较长。建议下载成功后,储存为 Rdata,以后方便使用。 | |
testdata <- GetAssayData (scRNA, slot="data") | |
clusters <- scRNA@meta.data$seurat_clusters | |
cellpred <- SingleR (test = testdata, ref = refdata, labels = refdata$label.fine, | |
# label.fine 耗时比较长一点 | |
method = "cluster", clusters = clusters, | |
assay.type.test = "logcounts", assay.type.ref = "logcounts") | |
save (cellpred,file = "../../tmp/cellpred.Rdata") | |
load ("../../tmp/cellpred.Rdata") | |
rm (refdata, HumanPrimaryCellAtlasData, testdata) #珍惜内存 | |
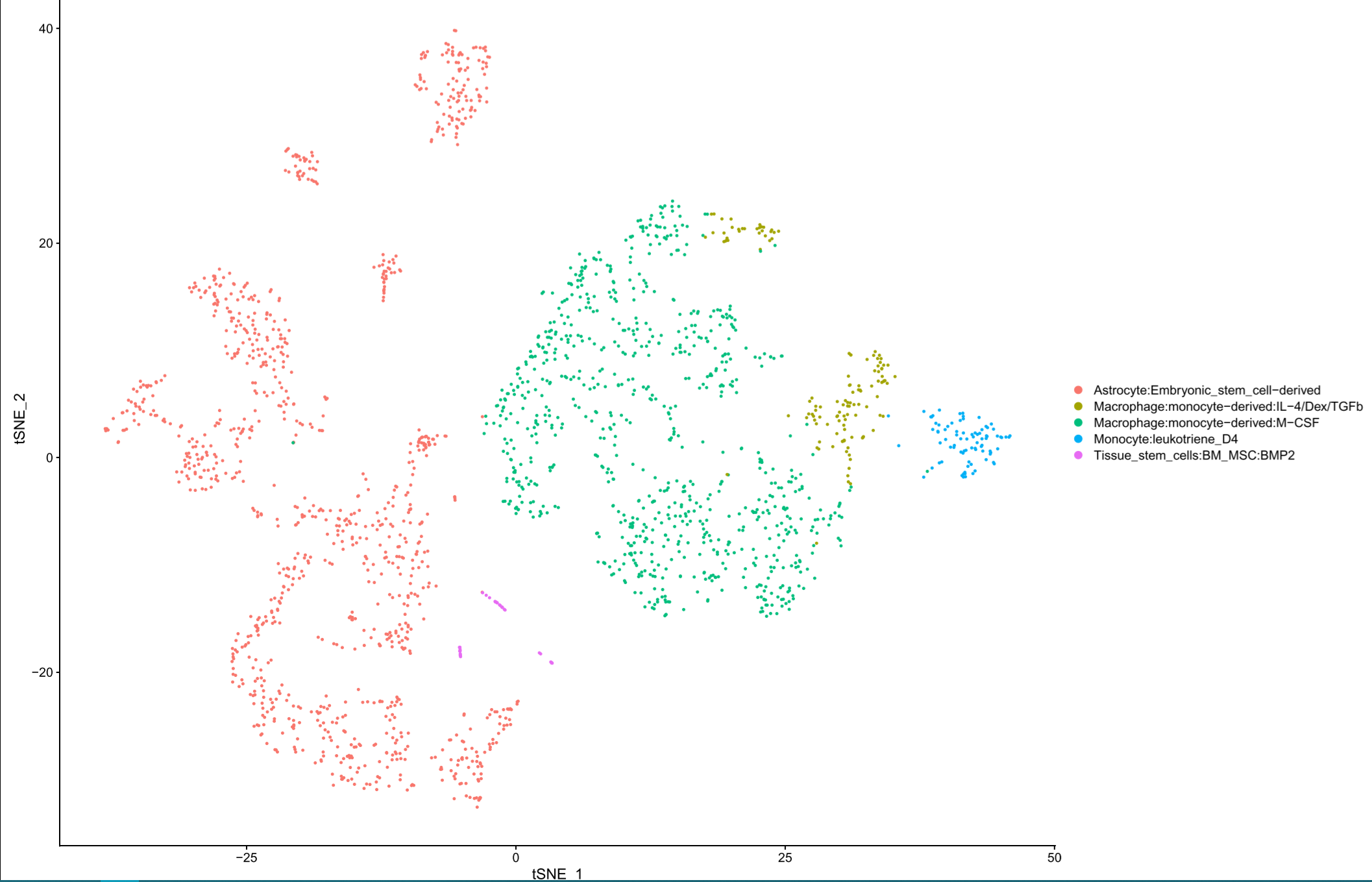
table (cellpred$labels) | |
celltype = data.frame (ClusterID=rownames (cellpred), celltype=cellpred$labels, stringsAsFactors = F) | |
table (celltype$ClusterID,celltype$celltype) #如下为 singleR 的细胞 cluster 鉴定结果。 | |
# 结合上述结果,给 scRNA 增添 celltype 注释信息 | |
scRNA@meta.data$celltype = "NA" | |
# 先新增列 celltype,值均为 NA,然后利用下一行代码循环填充 | |
for (i in 1:nrow (celltype)){ | |
scRNA@meta.data [which (scRNA@meta.data$seurat_clusters == celltype$ClusterID [i]),'celltype'] <- celltype$celltype [i]} | |
p1 <- DimPlot (scRNA, group.by="celltype", label=F , reduction='tsne') | |
ggsave ("../../out/3.3celltype_anno.pdf", plot = p1, width = 18, height = 12) |
<center>
细胞类型注释
</center>